Team: Mirja Thomsen (Postdoc), Annika Lange (intern), Frauke Hinrichs (technician), Linn Welzel (MD student), Katja Lohmann (group lead), Jonathan Krause (PhD student), Alexandra Rudnik (MD student), Martje Pauly (Clinician Scientist)
Our research team “Genetics of Rare Diseases” specializes in identifying pathogenic variants and genetic risk factors, focusing primarily on dystonia, Parkinson’s disease, ataxias, and neurodevelopmental disorders. We value national and international collaborations to identify rare and common genetic variants, combining clinical data with high-throughput genetic analyses, such as next-generation sequencing (exome, genome, transcriptome), to enhance early diagnosis and personalized treatments.
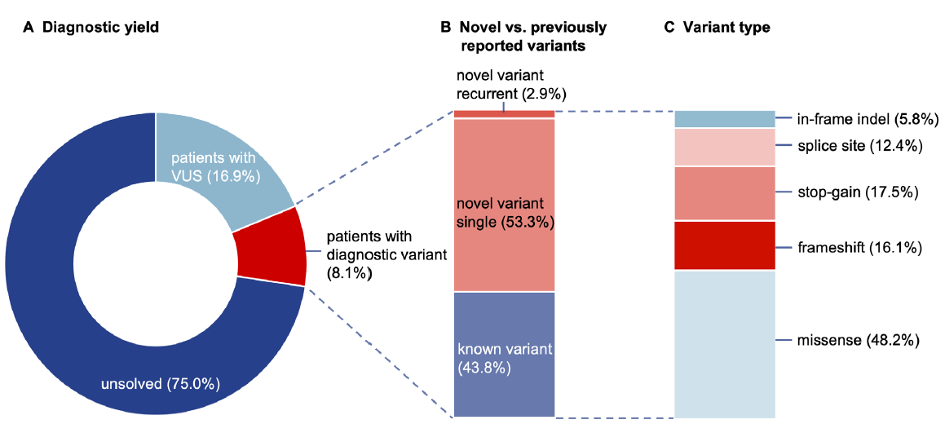
Our key achievements include the discovery of genetic risk factors for musician’s dystonia and novel disease genes, such as TUBB4A, RAB12, GNB1, VPS13D, and TAOK1. Furthermore, we contributed to the first descriptions of FGF14 and RFC1 repeat expansions in ataxia, as well as of pathogenic variants in EIF2AK2 and EIF4A2 in dystonia. Thousands of patients with Parkinson’s disease and dystonia from cohort studies and registers, including EPIPARK, DYSTRACT, and the Dystonia Coalition, have been screened using gene panels and microarray analyses, leading to the identification of mutation carriers who are being deeply clinically characterized.
Regarding rare diseases, we collaborate nationally and across Europe in initiatives such as T-NAMSE, Modellvorhaben Genomsequenzierung, and SolveRD, utilizing expert variant interpretation and sequencing techniques to enhance diagnosis and research. In the field of Parkinson´s disease, we participate in the large-scale international project Global Parkinson’s Genetics Program (GP2).
Significant contributions are also made to the MDSGene database, an international platform linked to the International Parkinson and Movement Disorder Society. It systematically collects and critically evaluates all known genetic variants related to movement disorders, serving as a comprehensive resource for clinicians and scientists on genotype-phenotype correlations and patient demographics.
Another major project in which we are involved is the ProtectMove study, which focuses on genetic risk and protective factors in movement disorders, particularly Parkinson’s disease and dystonia. We performed the genetic characterization of the ProMoveGene cohort, comprising approximately 15,000 individuals from diverse geographical backgrounds. We further have investigated genetic and epigenetic modifiers that affect penetrance and expressivity in monogenic movement disorders, using multiomics and systems biology approaches, with a focus on THAP1-related dystonia, genetic Parkinson´s disease, and ataxias, to better understand variable clinical outcomes among mutation carriers.
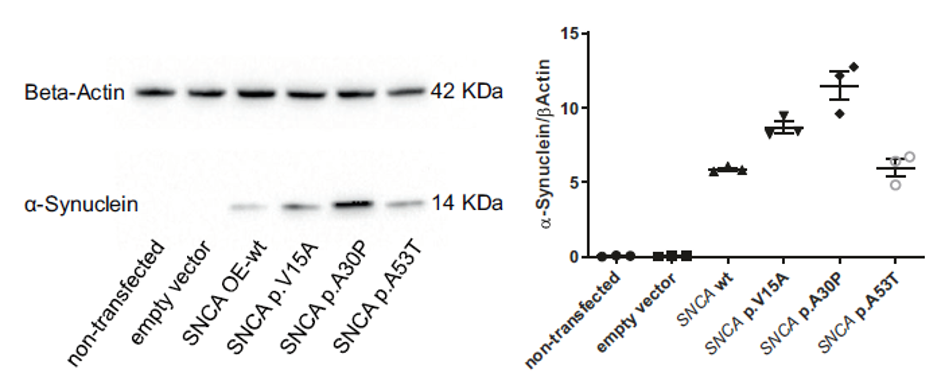
Further, we are also interested in understanding the molecular consequences of genetic variants in disease development and progression. To achieve this, we investigate selected molecular pathways through functional studies in cell and animal models, with the overarching aim of identifying targets for drug development.
Our team is internationally renowned for its work in human genetics, particularly in the field of rare neurological movement disorders, contributing to advancements in molecular diagnostics, global genetic databases, and the clinical application of genetic discoveries. This research is primarily funded by the German Research Foundation (DFG), the Federal Ministry of Education and Research (BMBF), the Dystonia Medical Research Foundation, and the Damp Foundation.